


Illustration by Paweł Jońca
In 2019, the Event Horizon Telescope team gave the world the first glimpse of what a black hole actually looks like. But the image of a glowing, ring-shaped object that the group unveiled wasn’t a conventional photograph. It was computed — a mathematical transformation of data captured by radio telescopes in the United States, Mexico, Chile, Spain and the South Pole1. The team released the programming code it used to accomplish that feat alongside the articles that documented its findings, so the scientific community could see — and build on — what it had done.
It’s an increasingly common pattern. From astronomy to zoology, behind every great scientific finding of the modern age, there is a computer. Michael Levitt, a computational biologist at Stanford University in California who won a share of the 2013 Nobel Prize in Chemistry for his work on computational strategies for modelling chemical structure, notes that today’s laptops have about 10,000 times the memory and clock speed that his lab-built computer had in 1967, when he began his prizewinning work. “We really do have quite phenomenal amounts of computing at our hands today,” he says. “Trouble is, it still requires thinking.”
Enter the scientist-coder. A powerful computer is useless without software capable of tackling research questions — and researchers who know how to write it and use it. “Research is now fundamentally connected to software,” says Neil Chue Hong, director of the Software Sustainability Institute, headquartered in Edinburgh, UK, an organization dedicated to improving the development and use of software in science. “It permeates every aspect of the conduct of research.”
Scientific discoveries rightly get top billing in the media. But Nature this week looks behind the scenes, at the key pieces of code that have transformed research over the past few decades.
Although no list like this can be definitive, we polled dozens of researchers over the past year to develop a diverse line-up of ten software tools that have had a big impact on the world of science. You can weigh in on our choices at the end of the story.
Language pioneer: the Fortran compiler (1957)
Contents
- 1 Language pioneer: the Fortran compiler (1957)
- 2 Signal processor: fast Fourier transform (1965)
- 3 Molecular cataloguers: biological databases (1965)
- 4 Forecast leader: the general circulation model (1969)
- 5 Number cruncher: BLAS (1979)
- 6 Microscopy must-have: NIH Image (1987)
- 7 Sequence searcher: BLAST (1990)
- 8 Preprint powerhouse: arXiv.org (1991)
- 9 Data explorer: IPython Notebook (2011)
- 10 Fast learner: AlexNet (2012)
The first modern computers weren’t user-friendly. Programming was literally done by hand, by connecting banks of circuits with wires. Subsequent machine and assembly languages allowed users to program computers in code, but both still required an intimate knowledge of the computer’s architecture, putting the languages out of reach of many scientists.
That changed in the 1950s with the development of symbolic languages — in particular the ‘formula translation’ language Fortran, developed by John Backus and his team at IBM in San Jose, California. Using Fortran, users could program computers using human-readable instructions, such as x = 3 + 5. A compiler then turned such directions into fast, efficient machine code.


This CDC 3600 computer, delivered in 1963 to the National Center for Atmospheric Research in Boulder, Colorado, was programmed with the help of the Fortran compiler.Credit: University Corporation for Atmospheric Research/Science Photo Library
It still wasn’t easy: in the early days, programmers used punch cards to input code, and a complex simulation might require tens of thousands of them. Still, says Syukuro Manabe, a climatologist at Princeton University in New Jersey, Fortran made programming accessible to researchers who weren’t computer scientists. “For the first time, we were able to program [the computer] by ourselves,” Manabe says. He and his colleagues used the language to develop one of the first successful climate models.
Now in its eighth decade, Fortran is still widely used in climate modelling, fluid dynamics, computational chemistry — any discipline that involves complex linear algebra and requires powerful computers to crunch numbers quickly. The resulting code is fast, and there are still plenty of programmers who know how to write it. Vintage Fortran code bases are still alive and kicking in labs and on supercomputers worldwide. “Old-time programmers knew what they were doing,” says Frank Giraldo, an applied mathematician and climate modeller at the Naval Postgraduate School in Monterey, California. “They were very mindful of memory, because they had so little of it.”
Signal processor: fast Fourier transform (1965)
When radioastronomers scan the sky, they capture a cacophony of complex signals changing with time. To understand the nature of those radio waves, they need to see what those signals look like as a function of frequency. A mathematical process called a Fourier transform allows researchers to do that. The problem is that it’s inefficient, requiring N2 calculations for a data set of size N.
In 1965, US mathematicians James Cooley and John Tukey worked out a way to accelerate the process. Using recursion, a ‘divide and conquer’ programming approach in which an algorithm repeatedly reapplies itself, the fast Fourier transform (FFT) simplifies the problem of computing a Fourier transform to just N log2(N) steps. The speed improves as N grows. For 1,000 points, the speed boost is about 100-fold; for 1 million points, it’s 50,000-fold.
The ‘discovery’ was actually a rediscovery — the German mathematician Carl Friedrich Gauss worked it out in 1805, but he never published it, says Nick Trefethen, a mathematician at the University of Oxford, UK. But Cooley and Tukey did, opening applications in digital signal processing, image analysis, structural biology and more. “It’s really one of the great events in applied mathematics and engineering,” Trefethen says. FFT has been implemented many times in code. One popular option is called FFTW, the ‘fastest Fourier transform in the west’.


A night view of part of the Murchison Widefield Array, a radio telescope in Western Australia that uses fast Fourier transforms for data collection.Credit: John Goldsmith/Celestial Visions
Paul Adams, who directs the molecular biophysics and integrated bioimaging division at Lawrence Berkeley National Laboratory in California, recalls that when he refined the structure of the bacterial protein GroEL in 19952, the calculation took “many, many hours, if not days”, even with the FFT and a supercomputer. “Trying to do those without the FFT, I don’t even know how we would have done that realistically,” he says. “It would have just taken forever.”
Molecular cataloguers: biological databases (1965)
Databases are such a seamless component of scientific research today that it can be easy to overlook the fact that they are driven by software. In the past few decades, these resources have ballooned in size and shaped many fields, but perhaps nowhere has that transformation been more dramatic than in biology.
Today’s massive genome and protein databases have their roots in the work of Margaret Dayhoff, a bioinformatics pioneer at the National Biomedical Research Foundation in Silver Spring, Maryland. In the early 1960s, as biologists worked to tease apart proteins’ amino acid sequences, Dayhoff began collating that information in search of clues into evolutionary relationships between different species. Her Atlas of Protein Sequence and Structure, first published in 1965 with three co-authors, described what was then known of the sequences, structures and similarities of 65 proteins. The collection was the first that “was not tied to a specific research question”, historian Bruno Strasser wrote in 20103. And it encoded its data in punch cards, which made it possible to expand the database and search it.
Other computerized biological databases followed. The Protein Data Bank, which today details more than 170,000 macromolecular structures, went live in 1971. Russell Doolittle, an evolutionary biologist at the University of California, San Diego, created another protein database called Newat in 1981. And 1982 saw the release of the database that would become GenBank, the DNA archive maintained by the US National Institutes of Health.
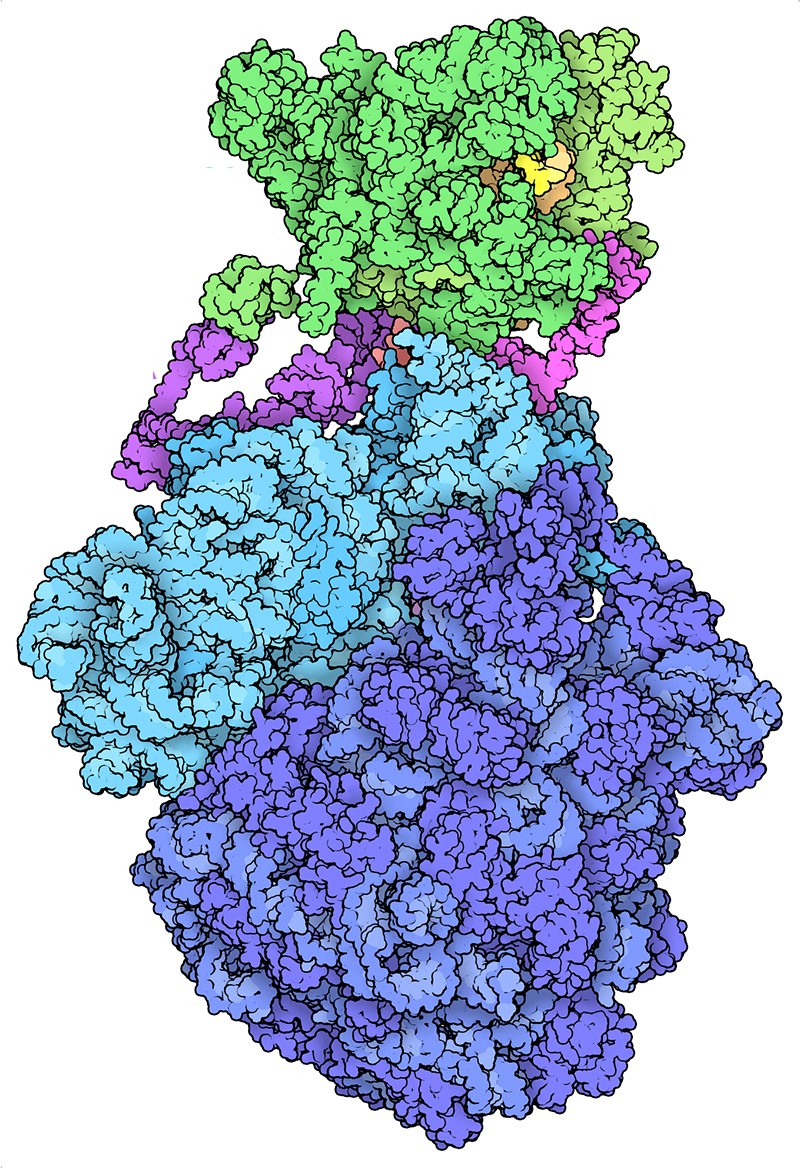

The Protein Data Bank has an archive of more than 170,000 molecular structures including this bacterial ‘expressome’, which combines the processes of RNA and protein synthesis.Credit: David S. Goodsell and RCSB PDB (CC BY 4.0)
Such resources proved their worth in July 1983, when separate teams led by Michael Waterfield, a protein biochemist at the Imperial Cancer Research Fund in London, and Doolittle independently reported a similarity between the sequences of a particular human growth factor and a protein in a virus that causes cancer in monkeys. The observation suggested a mechanism for oncogenesis-by-virus — that by mimicking a growth factor, the virus induces uncontrolled growth of cells4. “That set the light bulb off in some of the minds of biologists who were not into computers and statistics,” says James Ostell, former director of the US National Center for Biotechnology Information (NCBI): “We can understand something about cancer from comparing sequences.”
Beyond that, Ostell says, the discovery marked “an advent of objective biology”. In addition to designing experiments to test specific hypotheses, researchers could mine public data sets for connections that might never have occurred to those who actually collected the data. That power grows drastically when different data sets are linked together — something NCBI programmers achieved in 1991 with Entrez, a tool that allows researchers to freely navigate from DNA to protein to literature and back.
Stephen Sherry, current acting director of the NCBI in Bethesda, Maryland, used Entrez as a graduate student. “I remember at the time thinking it was magic,” he says.
Forecast leader: the general circulation model (1969)
At the close of the Second World War, computer pioneer John von Neumann began turning computers that a few years earlier had been calculating ballistics trajectories and weapon designs towards the problem of weather prediction. Up until that point, explains Manabe, “weather forecasting was just empirical”, using experience and hunches to predict what would happen next. Von Neumann’s team, by contrast, “attempted to do numerical weather prediction based upon laws of physics”.
The equations had been known for decades, says Venkatramani Balaji, head of the Modeling Systems Division at the National Oceanographic and Atmospheric Administration’s Geophysical Fluid Dynamics Laboratory in Princeton, New Jersey. But early meteorologists couldn’t solve them practically. To do so required inputting current conditions, calculating how they would change over a short time period, and repeating — a process so time-consuming that the mathematics couldn’t be completed before the weather itself caught up. In 1922, the mathematician Lewis Fry Richardson spent months crunching a six-hour forecast for Munich, Germany. The result, according to one history, was “wildly inaccurate”, including predictions that “could never occur under any known terrestrial conditions”. Computers made the problem tractable.
In the late 1940s, von Neumann established his weather-prediction team at the Institute for Advanced Study at Princeton. In 1955, a second team — the Geophysical Fluid Dynamics Laboratory — began work on what he called “the infinite forecast” — that is, climate modelling.
Manabe, who joined the climate modelling team in 1958, set to work on atmospheric models; his colleague Kirk Bryan addressed those for the ocean. In 1969, they successfully combined the two, creating what Nature in 2006 called a “milestone” in scientific computing.
Today’s models can divide the planet’s surface into squares measuring 25 × 25 kilometres, and the atmosphere into dozens of levels. By contrast, Manabe and Bryan’s combined ocean–atmosphere model5 used 500-km squares and 9 levels, and covered just one-sixth of the globe. Still, says Balaji, “that model did a great job”, allowing the team to test for the first time the impact of rising carbon dioxide levels in silico.
Number cruncher: BLAS (1979)
Scientific computing typically involves relatively simple mathematical operations using vectors and matrices. There are just a lot of them. But in the 1970s, there was no universally agreed set of computational tools for performing such operations. As a result, programmers working in science would spend their time devising efficient code to do basic mathematics rather than focusing on scientific questions.
What the programming world needed was a standard. In 1979, it got one: Basic Linear Algebra Subprograms, or BLAS6. The standard, which continued to evolve up to 1990, defined dozens of fundamental routines for vector and, later, matrix mathematics.
In effect, BLAS reduced matrix and vector mathematics to a basic unit of computation as fundamental as addition and subtraction, says Jack Dongarra, a computer scientist at the University of Tennessee in Knoxville who was a member of the BLAS development team.


Cray-1 supercomputer: before the BLAS programming tool was introduced in 1979, there was no linear algebra standard for researchers working on machines such as the Cray-1 supercomputer at Lawrence Livermore National Laboratory in California.Credit: Science History Images/Alamy
BLAS was “probably the most consequential interface to be defined for scientific computing”, says Robert van de Geijn, a computer scientist at the University of Texas at Austin. In addition to providing standardized names for common functions, researchers could be sure BLAS-based code would work in the same manner on any computer. The standard also enabled computer manufacturers to optimize BLAS implementations for speedy operation on their hardware.
More than 40 years on, BLAS represents the heart of the scientific computing stack, the code that makes scientific software tick. Lorena Barba, a mechanical and aerospace engineer at George Washington University in Washington DC, calls it “the machinery inside five layers of code”.
Says Dongarra, “It provides the fabric on which we do computing.”
Microscopy must-have: NIH Image (1987)
In the early 1980s, programmer Wayne Rasband was working with a brain-imaging lab at the US National Institutes of Health in Bethesda, Maryland. The team had a scanner to digitize X-ray films, but no way to display or analyse them on their computer. So Rasband wrote a program to do just that.
The program was specifically designed for a US$150,000 PDP-11 minicomputer — a rack-mounted, decidedly non-personal computer. Then, in 1987, Apple released its Macintosh II, a friendlier and much more affordable option. “It seemed obvious to me that that would work a lot better as a kind of laboratory image analysis system,” Rasband says. He ported his software to the new platform and rebranded it, seeding an image-analysis ecosystem.
NIH Image and its descendants empowered researchers to view and quantify just about any image, on any computer. The software family includes ImageJ, a Java-based version that Rasband wrote for Windows and Linux users, and Fiji, a distribution of ImageJ developed by Pavel Tomancak’s group at the Max Planck Institute of Molecular Cell Biology and Genetics in Dresden, Germany, that includes key plug-ins. “ImageJ is certainly the most foundational tool that we have,” says Beth Cimini, a computational biologist who works on the Imaging Platform of the Broad Institute in Cambridge, Massachusetts. “I’ve literally never spoken to a biologist who has used a microscope but not ImageJ or its offshoot project, Fiji.”


The ImageJ tool, with the help of a plug-in, can automatically identify cell nuclei in microscope images, as here.Credit: Ignacio Arganda-Carreras/ImageJ
That’s partly because these tools are free, Rasband says. But it’s also because it’s easy for users to customize the tool to their needs, says Kevin Eliceiri, a biomedical engineer at the University of Wisconsin–Madison, whose team has taken the lead on ImageJ development since Rasband’s retirement. ImageJ features a deceptively simple, minimalist user interface that has remained largely unchanged since the 1990s. Yet the tool is infinitely extensible thanks to its built-in macro recorder (which allows a user to save workflows by recording sequences of mouse clicks and menu selections), extensive file-format compatibility and flexible plug-in architecture. “Hundreds of people” have contributed plug-ins, says Curtis Rueden, the programming lead in Eliceiri’s group. These additions have greatly expanded the toolset for researchers, with functions to track objects over time in videos or automatically identify cells, for instance.
“The point of the program isn’t to be the be-all and end-all,” Eliceiri says, “it’s to serve the purpose of its users. And unlike Photoshop and other programs, ImageJ can be whatever you want it to be.”
Sequence searcher: BLAST (1990)
There might be no better indicator of cultural relevance than for a software name to become a verb. For search, think Google. And for genetics, think BLAST.
Evolutionary changes are etched into molecular sequences as substitutions, deletions, gaps and rearrangements. By searching for similarities between sequences — particularly among proteins — researchers can discover evolutionary relationships and gain insight into gene function. The trick is to do so quickly and comprehensively across rapidly ballooning databases of molecular information.
Dayhoff provided one crucial piece of the puzzle in 1978. She devised a ‘point accepted mutation’ matrix that allowed researchers to score the relatedness of two proteins based not only on how similar their sequences are, but also on the evolutionary distance between them.
In 1985, William Pearson at the University of Virginia in Charlottesville and David Lipman at the NCBI introduced FASTP, an algorithm that combined Dayhoff’s matrix with the ability to perform rapid searches.
Years later, Lipman, along with Warren Gish and Stephen Altschul at the NCBI, Webb Miller at Pennsylvania State University in University Park, and Gene Myers at the University of Arizona, Tucson, developed an even more powerful refinement: the Basic Local Alignment Search Tool (BLAST). Released in 1990, BLAST combined the search speed required to handle fast-growing databases with the ability to pick up matches that were more evolutionarily distant. At the same time, the tool could calculate how likely it is that those matches occurred by chance.
The result was incredibly fast, Altschul says. “You could put in your search, take one sip of coffee, and your search would be done.” But more importantly, it was easy to use. In an era when databases were updated by post, Gish established an e-mail system and later a web-based architecture that allowed users to run searches on the NCBI computers remotely, thus ensuring their results were always up-to-date.
The system gave the then-budding field of genome biology a transformative tool, says Sean Eddy, a computational biologist at Harvard University in Cambridge, Massachusetts — a way to work out what unknown genes might do on the basis of the genes they were related to. And for sequencing labs everywhere, it provided a clever neologism: “It’s just one of these things that became a verb,” Eddy says. “You just talked about BLASTing your sequences.”
Preprint powerhouse: arXiv.org (1991)
In the late 1980s, high-energy physicists routinely sent physical copies of their submitted manuscripts to colleagues by post for comment and as a courtesy — but only to a select few. “Those lower in the food chain relied on the beneficence of those on the A-list, and aspiring researchers at non-elite institutions were frequently out of the privileged loop entirely,” wrote physicist Paul Ginsparg in 20117.
In 1991, Ginsparg, then at Los Alamos National Laboratory in New Mexico, wrote an e-mail autoresponder to level the playing field. Subscribers received daily lists of preprints, each associated with an article identifier. With a single e-mail, users across the world could submit or retrieve an article from the lab’s computer system, get lists of new articles or search by author or title.
Ginsparg’s plan was to retain articles for three months, and to limit content to the high-energy physics community. But a colleague convinced him to retain the articles indefinitely. “That was the moment it transitioned from bulletin board to archive,” he says. And papers flooded in from much farther afield than Ginsparg’s own discipline. In 1993, Ginsparg migrated the system to the Internet, and in 1998 he gave it the name it goes by today: arXiv.org.
Now in its thirtieth year, arXiv houses some 1.8 million preprints — all available for free — and attracts more than 15,000 submissions and some 30 million downloads per month. “It’s not hard to see why the arXiv is such a popular service,” the editors of Nature Photonics wrote8 a decade ago on the occasion of the site’s twentieth anniversary: “The system provides researchers with a fast and convenient way to plant a flag that shows what they did and when, avoiding the hassle and time required for peer review at a conventional journal.”
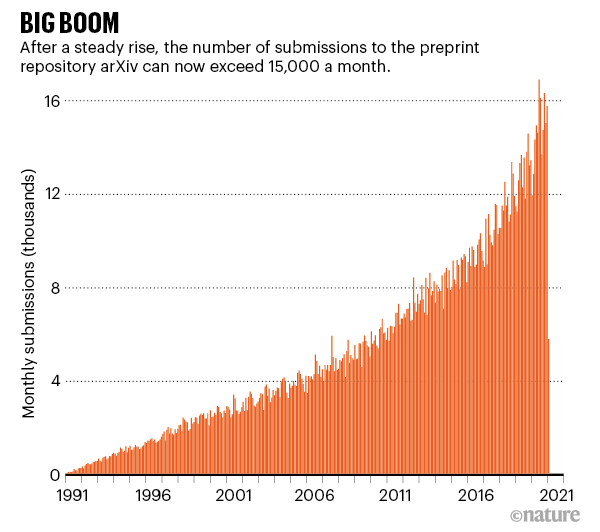

Source: arXiv.org
The site’s success catalysed a boom in sister archives in biology, medicine, sociology and other disciplines. The impact can be seen today in tens of thousands of preprints that have been published on the virus SARS-CoV-2.
“It’s gratifying to see a methodology, considered heterodox outside of the particle-physics community 30 years ago, now more generally viewed as obvious and natural,” Ginsparg says. “In that sense, it’s like a successful research project.”
Data explorer: IPython Notebook (2011)
Fernando Pérez was a graduate student “in search of procrastination” in 2001 when he decided to take on a core component of Python.
Python is an interpreted language, which means programs are executed line by line. Programmers can use a kind of computational call-and-response tool called a read–evaluate–print loop (REPL), in which they type code and a program called an interpreter executes it. A REPL allows for quick exploration and iteration, but Pérez noted that Python’s wasn’t built for science. It didn’t allow users to easily preload modules of code, for instance, or keep data visualizations open. So Pérez wrote his own version.
The result was IPython, an ‘interactive’ Python interpreter that Pérez unveiled in December 2001 — all 259 lines of it. A decade later, Pérez, working with physicist Brian Granger and mathematician Evan Patterson, migrated that tool to the web browser, launching the IPython Notebook and kick-starting a data-science revolution.
Like other computational notebooks, IPython Notebook combined code, results, graphics and text in a single document. But unlike other such projects, IPython Notebook was open-source, inviting contributions from a vast developer community. And it supported Python, a popular language for scientists. In 2014, IPython evolved into Project Jupyter, supporting some 100 languages and allowing users to explore data on remote supercomputers as easily as on their own laptops.
“For data scientists, Jupyter has emerged as a de facto standard,” Nature wrote in 20189. At the time, there were 2.5 million Jupyter notebooks on the GitHub code-sharing platform; today, there are nearly 10 million, including the ones that document the 2016 discovery of gravitational waves and the 2019 imaging of a black hole. “That we made a small contribution to those projects is extremely rewarding,” Pérez says.
Fast learner: AlexNet (2012)
Artificial intelligence (AI) comes in two flavours. One uses codified rules, the other enables a computer to ‘learn’ by emulating the neural structure of the brain. For decades, says Geoffrey Hinton, a computer scientist at the University of Toronto, Canada, AI researchers dismissed the latter approach as “nonsense”. In 2012, Hinton’s graduate students Alex Krizhevsky and Ilya Sutskever proved otherwise.
The venue was ImageNet, an annual competition that challenges researchers to train an AI on a database of one million images of everyday objects, then test the resulting algorithm on a separate image set. At the time, the best algorithms miscategorized about one-quarter of them, Hinton says. Krizhevsky and Sutskever’s AlexNet, a ‘deep-learning’ algorithm based on neural networks, reduced that error rate to 16%10. “We basically halved the error rate, or almost halved it,” notes Hinton.
Hinton says the team’s success in 2012 reflected the combination of a big-enough training data set, great programming and the newly emergent power of graphical processing units — the processors that were originally designed to accelerate computer video performance. “Suddenly we could run [the algorithm] 30 times faster,” he says, “or learn on 30 times as much data.”
The real algorithmic breakthrough, Hinton says, actually occurred three years earlier, when his lab created a neural network that could recognize speech more accurately than could conventional AIs that had been refined over decades. “It was only slightly better,” Hinton says. “But already that was the writing on the wall.”
Those victories heralded the rise of deep learning in the lab, the clinic and more. They’re why mobile phones are able to understand spoken queries and why image-analysis tools can readily pick out cells in photomicrographs. And they are why AlexNet takes its place among the many tools that have fundamentally transformed science, and with them, the world.
